Published on
October 26, 2025


Regulatory due diligence in biotech is critical for securing funding and navigating complex compliance requirements. Investors focus on three main areas: FDA approval status, clinical trial compliance, and quality systems like GMP/GLP. To meet these expectations, biotech companies must maintain thorough documentation, address potential red flags, and demonstrate readiness to handle regulatory challenges.
Key steps include:
Proactive preparation reduces risks, builds investor confidence, and supports funding and M&A efforts. Advisory services can help streamline this process, aligning regulatory milestones with financial strategies.
When it comes to regulatory due diligence, having well-organized documentation and a centralized approach to data management is non-negotiable. By categorizing core documents into three key areas, you can set the stage for handling regulatory hurdles effectively.
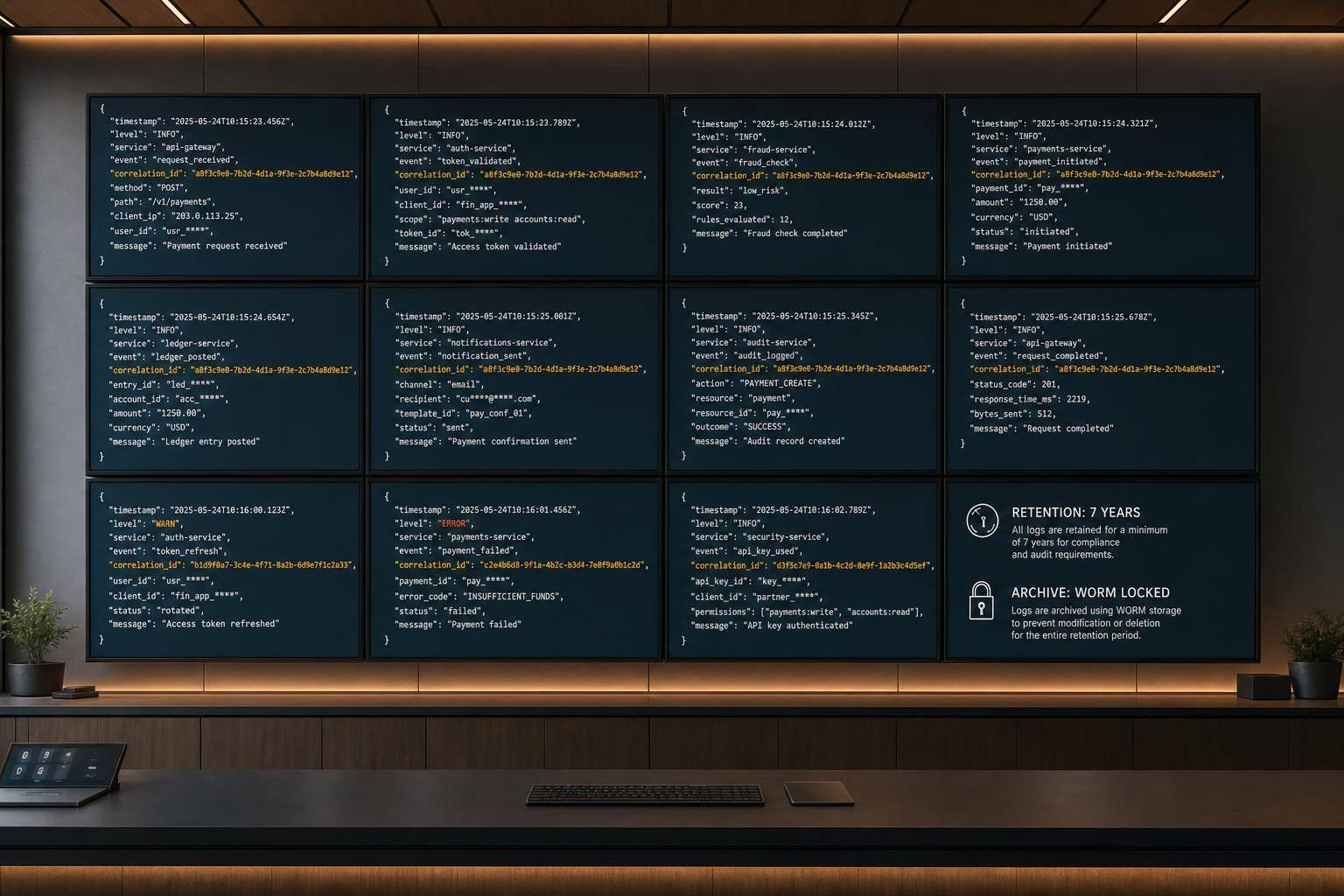
Your Investigational New Drug (IND) applications are the backbone of your FDA submission portfolio. These files need to be thorough, up-to-date, and readily accessible in your virtual data room. Make sure to include original filings, amendments, and safety reports to showcase compliance with FDA standards.
If your company has moved to the next phase, New Drug Applications (NDA) and Biologics License Applications (BLA) become critical. Organize these documents in chronological order, ensuring each submission includes complete application packages, responses from the FDA, and any required modifications. Use the MM/DD/YYYY format for labeling submission dates, aligning with U.S. regulatory norms.
Another area of focus is FDA correspondence, which is often scrutinized by investors. Gather all meeting minutes, official communications, and feedback from FDA interactions. This should cover pre-IND meeting summaries, records from Type A, B, and C meetings, and informal exchanges with FDA staff. These documents provide a clear picture of your regulatory approach and the FDA's input.
After every interaction with the FDA, perform a thorough audit of your records. Keeping everything current eliminates last-minute stress during due diligence and highlights your operational discipline to potential investors.
Managing clinical trial records with precision is just as important as FDA documentation.
Start by organizing trial protocols, including version-controlled amendments, detailed statistical plans, and Institutional Review Board (IRB) approvals with renewal schedules. These materials serve as the foundation of your clinical trial documentation.
Next, ensure Good Clinical Practice (GCP) compliance records are in order. This includes GCP training records for your team, monitoring reports from clinical research organizations, and audit findings along with corrective actions. These documents underscore your adherence to ethical and scientifically rigorous research practices.
For informed consent forms, organize them by study and site, ensuring they include dated approvals and an update log.
Finally, clinical trial results and data integrity should be well-documented. Provide complete datasets, statistical analysis reports, and third-party audits of your clinical data. Include data management plans and proof of reliable data collection systems to reassure investors about the credibility of your findings.
A well-structured patent portfolio is essential for giving investors a clear view of your intellectual property strategy. Create a summary table that lists all patents, along with their status, filing dates, numbers, and key claims. Keep this document updated and cross-referenced with detailed patent files.
Freedom to operate analyses are another critical piece. Include legal opinions that assess potential IP conflicts and evaluate your ability to commercialize your products without infringing on existing patents. These should cover both U.S. and international markets relevant to your goals.
Organize all licensing agreements and IP assignments carefully. Maintain comprehensive records of licensing deals, including financial terms, milestone obligations, and restrictions on your freedom to operate. Ensure IP ownership records are clear and readily available for review.
Transparency is key when it comes to litigation and IP challenges. If your company is involved in any patent disputes or freedom to operate issues, provide full legal files, expert opinions, and strategic assessments of potential outcomes. Being upfront about these matters builds trust with investors and shows that your team is prepared to handle IP risks effectively.
For expert assistance in managing your regulatory documentation, consider working with advisory groups like Phoenix Strategy Group. They offer specialized services to help you organize and maintain compliance records, ensuring you're ready for investor scrutiny. Their data engineering solutions can also establish systems to keep your records updated throughout your development process.
Investing in proper documentation organization isn’t just about securing immediate funding. It simplifies future FDA interactions, streamlines mergers and acquisitions, and demonstrates the operational maturity that seasoned investors expect from biotech companies.
Securing investor confidence often hinges on how well you address key regulatory areas. Clear and thorough documentation not only showcases your preparedness but also strengthens your case during funding evaluations.
Navigating FDA approval pathways involves understanding their unique requirements, timelines, and documentation needs.
For medical devices, the 510(k) pathway typically takes 1–3 years and focuses on proving substantial equivalence to an already-approved device. The Premarket Approval (PMA) process, however, applies to more complex devices and can take 3–7 years. On the pharmaceutical side, New Drug Applications (NDA) and Biologics License Applications (BLA) represent the most stringent pathways, also requiring 3–7 years to complete.
Your submission package should include detailed predicate device comparisons, clinical study summaries, full clinical trial data, manufacturing processes, and a well-developed risk-benefit analysis. Organize all records in chronological order using the MM/DD/YYYY format, which aligns with U.S. regulatory standards. Ensure that every stage of development is documented, from IND submissions to clinical trial outcomes and scale-up manufacturing details.
Consider creating a pathway status dashboard to visually map your progress, upcoming milestones, and realistic timelines. This tool can help demonstrate how your strategy aligns with regulatory expectations and provide clarity for both investors and regulators.
Next, ensure your clinical trial processes and data management systems meet regulatory standards.
Once your trial records are in order, shift focus to ensuring Good Clinical Practice (GCP) compliance and robust adverse event reporting systems. GCP compliance is the foundation of clinical trial regulation and reflects your ability to meet regulatory demands.
Adverse event reporting is another critical area. Clearly document your procedures for collecting adverse event data, your timelines for reporting to the FDA, and your safety monitoring protocols. Investors will likely benchmark your performance against industry standards, so addressing any gaps proactively is essential.
Your data management plans should reflect strong systems for collecting, storing, and analyzing clinical data. Include details on data validation, audit trails, and backup processes to reassure investors that your data can withstand FDA scrutiny.
Organize clinical monitoring reports from contract research organizations (CROs) by study and site. Highlight any corrective actions taken and their resolution status to demonstrate operational efficiency and readiness for regulatory review.
Compliance with Good Manufacturing Practice (GMP) and Good Laboratory Practice (GLP) standards is critical to proving you can produce safe, effective products at scale.
Maintain up-to-date Standard Operating Procedures (SOPs) for equipment calibration, personnel training, batch record management, and quality control testing. Include version control logs and training records to show that your team is prepared and capable.
Both internal and external audit reports are essential for evaluating your quality systems. Document all findings, corrective action plans, and resolution timelines. Follow up with additional audits to confirm that corrective measures have been successfully implemented. Annual GMP/GLP audits are typical, so ensure your schedule meets or exceeds these expectations.
Your quality assurance records should cover equipment qualification, facility validation, and environmental monitoring. For temperature-sensitive products, maintain continuous monitoring records in Fahrenheit to comply with U.S. standards. Include calibration certificates and validation protocols for all critical equipment and manufacturing processes.
Keep comprehensive personnel training documentation to demonstrate a strong quality culture. Track initial certifications, annual refresher courses, and any supplementary training required when procedures are updated. Competency assessments should also be included to confirm staff readiness.
Implement a compliance tracking system to monitor quality metrics, audit schedules, and training statuses in real time. This approach not only reassures investors but also embeds quality management deeply within your organization.
For additional support, consider working with advisory firms like Phoenix Strategy Group. They can help streamline compliance documentation, establish tracking systems, and assist with budgeting for ongoing regulatory requirements while ensuring your records remain accessible throughout the due diligence process.
Regulatory missteps can delay biotech funding by over 50%. Recognizing these warning signs and addressing them with clear strategies can make the difference between securing funding and losing investor trust. Let’s dive into key red flags and how to tackle them effectively.
Incomplete or inconsistent FDA submissions are a major red flag. If your Investigational New Drug (IND) application history shows gaps or your FDA correspondence lacks clarity, investors may question your regulatory preparedness and overall risk level.
To avoid this, conduct a thorough audit of your FDA submissions before entering due diligence. Create a detailed timeline (using the MM/DD/YYYY format) that captures every FDA interaction, from initial meetings to the latest status updates. Double-check that all documentation is complete, properly formatted, and meets FDA standards.
Engaging regulatory consultants early can be a game-changer. These experts can identify discrepancies that your internal team might miss, flagging issues and suggesting corrections before they become a problem.
Once your FDA filings are in order, focus on clinical trial data integrity to further build confidence.
Clinical trial data problems - like missing source data, incomplete patient records, or unexplained protocol deviations - can quickly derail funding conversations.
To prevent these issues, implement strong data management practices and third-party verification systems. Every data point should have a clear source, proper documentation, and validation records. Address any inconsistent or missing data proactively, rather than waiting for investors to uncover them. For protocol deviations, provide detailed explanations, including the reasoning behind changes and their impact on study outcomes. Document all corrective actions and resolutions thoroughly.
Keep adverse event records complete and up-to-date, including reporting timelines and internal safety monitoring protocols.
Mock audits conducted by external consultants can be incredibly valuable. They allow you to identify and resolve data issues privately, demonstrating to investors that you prioritize data integrity and transparency.
With clinical data squared away, it’s time to focus on intellectual property (IP) concerns, which are equally critical.
Weak IP protection can shake investor confidence, especially if your core technology lacks strong patent coverage or faces unresolved disputes. Warning signs include contested patents, incomplete freedom-to-operate analyses, and unclear ownership documentation for inventions.
Start by reviewing your entire patent portfolio, including pending applications and granted patents in key markets. Ensure all patents are up to date, with fees paid and renewal dates tracked. Clearly document the scope of protection each patent provides and how it aligns with your commercial strategy.
Unresolved IP disputes are a red flag you can’t afford. Work with experienced IP counsel to resolve these issues through settlement, licensing agreements, or alternative dispute resolution methods. Investors need to see certainty in your IP position.
Strengthen your freedom-to-operate by analyzing third-party patents that could impact your commercialization plans. Document your findings, along with any licensing agreements or design-around strategies you’ve implemented.
Review all invention assignment agreements with employees, contractors, and collaborators to ensure clear ownership of your IP assets. Missing or unclear agreements can lead to disputes that complicate funding or acquisition efforts.
If your portfolio has weak spots, consider filing additional patent applications. Continuation applications or international filings can expand your protection and demonstrate ongoing innovation.
For companies preparing for regulatory due diligence, working with specialized advisors like Phoenix Strategy Group can be invaluable. Their expertise in financial planning, data management, and M&A support helps biotech companies address these red flags, implement compliance systems, and reassure investors - all while keeping the focus on development goals.
Tackling regulatory due diligence on your own can feel like an uphill battle. Expert advisory support can turn this daunting process into an advantage, helping biotech companies showcase their operational strengths while staying focused on their primary goal: developing life-saving treatments. This type of guidance not only boosts readiness for regulatory reviews but also strengthens appeal to investors.
A skilled advisory partner brings expertise in areas like financial modeling, data management, and regulatory compliance - skills that biotech teams often lack in-house. This support becomes indispensable during funding rounds or M&A transactions, where regulatory preparedness can make or break a deal.
Reaching regulatory milestones requires substantial financial resources, often with unpredictable costs and timelines. Milestone-based financial modeling helps biotech companies align their cash flow with these demands, ensuring they can navigate critical stages such as IND submissions, Phase II trials, and FDA approvals without financial strain.
Phoenix Strategy Group has proven success in this area, helping portfolio companies secure over $200 million in funding within the last year. Their fractional CFO services focus on strategic finance, forecasting, and budgeting, all tailored to regulatory timelines.
Developing scenario-based cash flow models with real-time variance analysis dashboards not only keeps expenses on track but also builds trust with investors.
"As our fractional CFO, they accomplished more in six months than our last two full-time CFOs combined. If you're looking for unparalleled financial strategy and integration, hiring PSG is one of the best decisions you can make." - David Darmstandler, Co-CEO, DataPath
By aligning budgets with key regulatory milestones, companies can ensure adequate funding for each phase while maintaining flexibility for unexpected challenges. Beyond financial planning, maintaining reliable and accessible data is equally critical for compliance.
Staying on top of compliance is non-negotiable when it comes to regulatory readiness. Real-time compliance tracking, supported by advanced data engineering, allows biotech companies to monitor FDA submission timelines, clinical trial enrollment rates, adverse event reporting, and GMP/GLP audit outcomes. Centralizing this data through ETL pipelines and data warehousing ensures it’s validated and easily accessible during due diligence.
Cloud-based data lakes and automated compliance systems reduce manual errors and provide the transparency investors expect. Advanced analytics can even help predict compliance issues before they arise, enabling proactive solutions.
Integrating these systems with existing platforms - like clinical trial management or quality management systems - ensures seamless data flow. This not only safeguards data integrity but also reduces the administrative load on internal teams.
Companies leveraging advanced data management tools for compliance tracking stand out during investor evaluations. A strong data management framework shows operational excellence and supports effective fundraising and M&A strategies.
The biotech industry’s regulatory complexities can complicate fundraising and M&A efforts. Expert advisory services help companies structure deals, time funding rounds around regulatory milestones, and prepare documentation that satisfies both investors and regulatory bodies.
Phoenix Strategy Group brings decades of experience to the table, having completed over 100 M&A transactions and supported more than five IPOs since 1998. Their investment banking services include valuation, due diligence systems, deal structuring, and negotiation support - all tailored to regulated industries.
Timing is everything when aligning fundraising with regulatory events. Companies that secure funding shortly before or after favorable FDA interactions often achieve better valuations and terms. Advisory partners help pinpoint these timing windows while ensuring all regulatory documents are ready for investor review.
The advisory process also focuses on creating robust due diligence systems that anticipate investor questions, streamline reviews, and minimize the risk of regulatory red flags. Advisors assist with structuring milestone-based payments, regulatory contingencies, and intellectual property protections, ensuring consistency between financial and regulatory strategies.
For biotech firms gearing up for major funding rounds or exit opportunities, Phoenix Strategy Group offers the financial, data management, and strategic expertise needed to navigate regulatory challenges and maximize enterprise value.
Regulatory due diligence in biotech isn’t just a box to check - it’s a crucial factor that can make or break your funding efforts. With the biotech sector recently drawing in a staggering $65 billion in funding, competition among startups is fierce. Only those with airtight regulatory preparation stand out to investors and move forward in the funding process.
At the heart of successful due diligence lies comprehensive documentation. This includes FDA submissions, clinical trial records, and intellectual property (IP) filings presented in a clear, organized manner. Missing or incomplete documents are one of the biggest red flags for investors, often derailing funding opportunities. By maintaining meticulous records, you not only reduce investor concerns but also create a strong foundation for resolving any potential issues.
Investors often focus on three key areas:
These factors directly influence your company’s risk profile, timeline to market, and ultimately, its valuation - critical elements for investment decisions.
Beyond documentation, taking proactive steps to address risks boosts investor confidence. Closing gaps in FDA filings, resolving clinical trial data inconsistencies, and fortifying IP protections demonstrate a level of operational readiness that reassures investors about your ability to navigate regulatory challenges.
Expert advisory support plays a pivotal role here. Companies that tap into specialized expertise in regulatory, financial, and data management consistently outperform those trying to handle due diligence alone. This guidance becomes especially valuable for milestone-based financial planning, compliance tracking, and preparation for mergers and acquisitions.
Investors also pay attention to advanced compliance tracking systems. Real-time monitoring of FDA submission timelines, clinical trial progress, and audit results not only ensures you’re ready for regulatory scrutiny but also highlights your operational efficiency - an attractive quality for potential backers.
Strategic timing is another critical element. Aligning funding efforts with major regulatory milestones - such as successful IND (Investigational New Drug) submissions or positive Phase II trial results - can lead to better valuations and more favorable investment terms. This requires careful coordination between regulatory achievements and financial planning.
For biotech founders, investing in thorough regulatory due diligence doesn’t just secure funding - it lays the groundwork for growth, builds trust with investors, and positions your company for long-term success.
When putting together regulatory documentation for investor review, biotech companies need to focus on a few critical elements to make the due diligence process as seamless as possible:
Careful preparation not only reassures investors but also underscores your company’s dedication to meeting regulatory expectations. For extra guidance, you might want to collaborate with advisors who specialize in helping growth-stage companies navigate regulatory and financial readiness.
To handle potential red flags during regulatory due diligence, biotech companies need to take a proactive and meticulous approach. Start by performing an internal review of all regulatory filings, approvals, and compliance records. This ensures everything is accurate and current. If you spot any discrepancies or missing information, address them well before presenting to potential investors.
Make sure to have detailed documentation ready for critical areas like clinical trial data, intellectual property protections, and adherence to FDA or other relevant regulatory requirements. Showing investors that you fully understand your regulatory obligations - and have a plan to tackle any challenges - can go a long way in building their trust.
It’s also a good idea to bring in seasoned advisors or consultants with expertise in regulatory due diligence. Their insights can help you identify risks early on and improve your preparation. These steps not only reduce concerns but also position your company as a reliable and ready investment opportunity.
Aligning funding strategies with regulatory milestones is a smart move for biotech companies. Why? Because it ensures you’ve got the financial backing you need during those pivotal moments in your product’s development - like clinical trials or FDA approvals. These processes aren’t just costly; they’re also highly time-sensitive. Proper planning keeps everything on track, reassures investors, and shows you’ve got a solid plan to hit your key objectives.
Here’s how to make it happen: Start by creating a detailed timeline of your regulatory milestones. Then, map out the funding requirements for each phase. It’s also important to keep open lines of communication with stakeholders and prioritize proactive financial planning. Need extra support? Partnering with experts like Phoenix Strategy Group can help you fine-tune your financial strategies, so you’re ready to tackle both regulatory and funding hurdles with confidence.